Kronikk: Arvid Heiberg
Genterapi for sjeldne sykdommer
Michael 2021; 18: 83–90.
Ved genterapi endres gener eller genuttrykk i terapeutisk øyemed. Innen feltet sjeldne sykdommer har det siden 1990 vært mange forsøk, men få konkrete resultater. Dette er i ferd med å endre seg. Syv genterapeutiske medikamenter for sjeldne sykdommer er godkjent (2020). Vi har fått metoder som gjør genterapi og genredigering mulig, men det er problemer ved å ta slike medikamenter og metoder i bruk, blant annet kostnadene.
Genterapi består i å endre kroppens gener eller deres uttrykk. I medisinsk sammenheng gjør vi dette for å kurere eller mildne sykdom. Da må man enten reparere eller komplettere den genetiske effekten i celler eller vev der sykdommen utspiller seg, eller gi cellen en ny funksjon. Til å overføre eller undertrykke disse genene, eller deler av disse, bruker man vektorer, også kalt genbærere. Det er vanligvis svekkede virus som er ufarlige for mennesker fordi de ikke lenger gir sykdom.
Interessen for genterapi har lenge vært til stede, men fordi terapien har gått fra å være lovende på sikt, men i praksis mislykket flere ganger, avtok optimismen. Nå har man imidlertid igjen økende optimisme på en rekke felt (1-4).
Presisjonsmedisin eller persontilpasset medisin har to mål: Det ene er å finne riktig medisin for den spesifikke pasienten. Det andre, mer indirekte målet, er å ikke behandle enkeltpasienter med medikamenter som har liten eller ingen virkning.
Det norske programmet for persontilpasset medisin har tre satsningsområder: kreft (4), mikrobiologi og sjeldne sykdommer. I denne kronikken vil jeg omtale de sjeldne sykdommene og utvikling av genterapien på dette feltet.
Genterapi ved sjeldne sykdommer
Innen feltet genterapi ved sjeldne sykdommer har det siden 1990 vært mange forsøk, oversalg og feilslag. Det har vært en rekke etiske bekymringer med hensyn til sikkerhet, smittsomhet, varighet og effekt, foruten risiko for at man kan overføre egenskaper til neste generasjon. Imidlertid er feltet modnet og bekymringene mindre, men til dels fortsatt til stede. Det er nå syv godkjente medikamenter på markedet i USA og Europa.
Antall artikler på stikkordet gene therapy i litteraturdatabasen Pubmed økte fra rundt 7 000 i 2000 til nær 30 000 i 2019. Tilsvarende oppslag i clinicaltrials.gov, en database for kliniske studier, viser at det er registrert 631 forsøk på rare diseases (11.10.2020). Dette er alt fra avsluttede til pågående forsøk og gjelder alle organsystemer. Det er enda større aktivitet innen kreftforskning, noe som kan komme sjeldenfeltet til nytte.
Globalt antar man at genterapiforsøk på mange tusen pasienter med sjeldne sykdommer har vært gjennomført eller pågår (1). Mange fase 3-forsøk foregår og vil etter alt å dømme gi preparater i praktisk bruk de kommende år.
De første vellykkede forsøk skjedde i 1990 og var rettet mot den sjeldne sykdommen X-bundet SCID (severe combined immuno-deficiency av X-nedarvet type) (1). Men samtidig var dessverre datidens virusvektorer i stand til å aktivere pasientens latente onkogener (kreftdisponerende gener), slik at man avsluttet forsøkene og startet jakten på bedre vektorer. Nå er det oftest adenoassosierte virus med lav patogenisitet hos mennesker som benyttes, ved at man pakker genet eller genmaterialet inn i vektoren som så infiserer relevant vev og tvinger cellen til å produsere «riktig genmateriale».
Genredigering
Nobelprisen i kjemi for 2020 ble tildelt forskerne Emmanuelle Charpentier og Jennifer A. Doudna for deres oppdagelse av CRISPR (clustered regularly interspaced short palindromic repeats). Systemet er kjent fra bakteriers forsvar mot virusinfeksjoner, men sammen med Feng Zhang fant forskerne i 2012 at dette kunne brukes til å endre DNA i celler og vev hos pattedyr (2, 3).
Ved CRISPR-systemet sammen med Cas9 (CRISPR-assosiert protein 9) kan man klippe ut biter med mutasjoner i genet og sette inn den riktige sekvensen (figur 1) (2). Metoden er relativt enkel og billig. Man kan kjøpe reagensene for et par hundrelapper. Med ferdige kits kan man «ordne seg på kjøkkenet» (3). Metoden blir ofte kalt «klipp-og-lim»-teknikk. Fordelen med denne i forhold til tidligere brukte metoder, er at man har vesentlig bedre kontroll på hvor og hva man klipper ut og på hva man setter inn. Det er lettere å inaktivere gener enn å sette inn korrekte genbiter, og man kan derfor bruke dette til å skru av gener som man vet har skadelig effekt. Metoden er rapportert vellykket brukt på en pasient med sigdcelleanemi (5). Den er også brukt til å modifisere pasientens egen hud, gro reparert hud i laboratoriet og så transplantere dette tilbake ved en hudsykdom med alvorlig blæredannelse (junksjonal epidermolysis bullosa) (6).
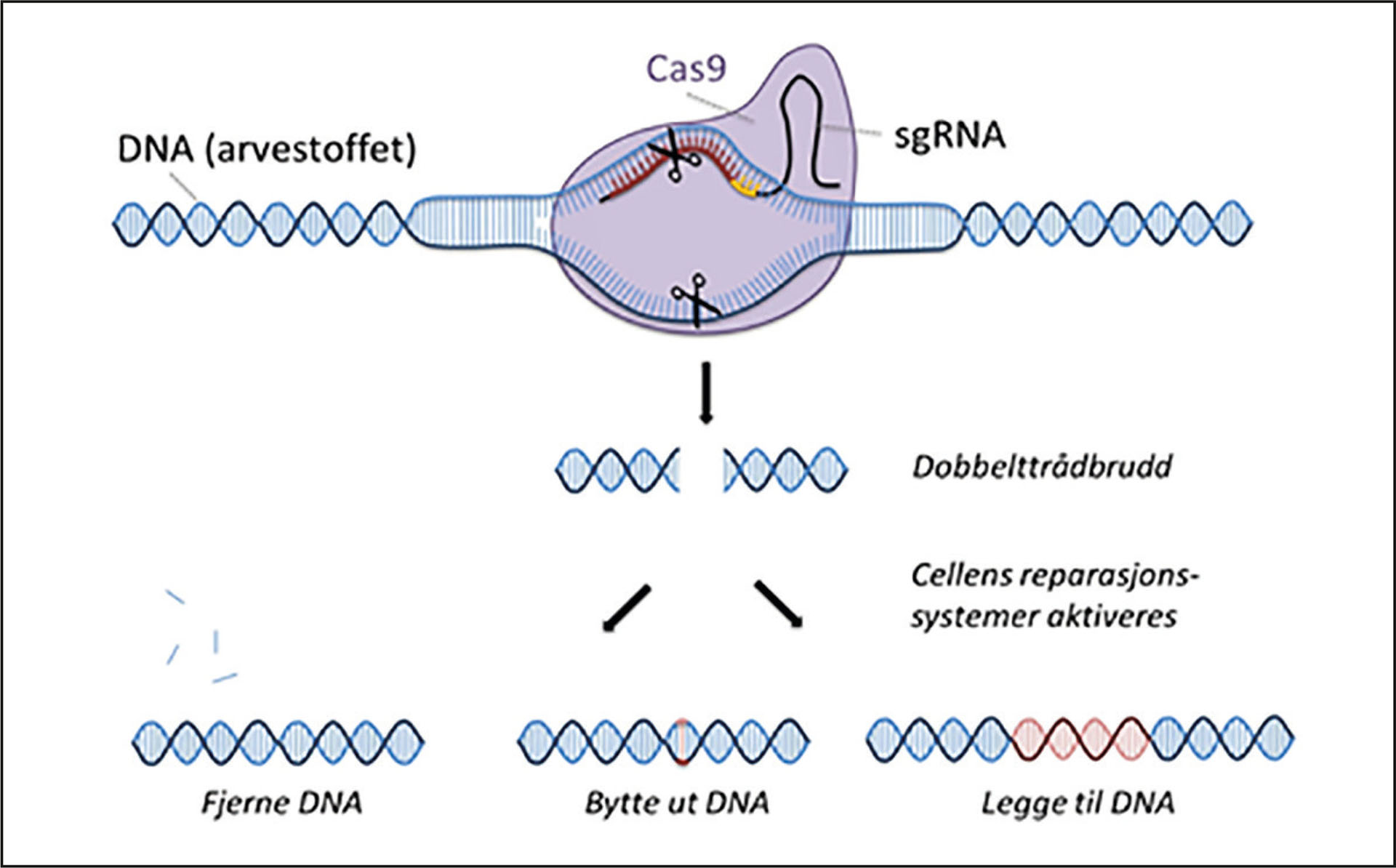
Figur 1: Ved genredigering med CRISPR kuttes DNA-et av et enzym kalt Cas9. Det er et spesielt RNA-molekyl (kalt sgRNA) som bestemmer hvor kuttet lages ved å binde seg til en matchende sekvens i DNA-et. Man kan derfor selv bestemme hvor DNA-et skal kuttes. Cellen liker ikke kuttet DNA, så den iverksetter systemer for å reparere det. Ved å manipulere denne prosessen kan man ta vekk, bytte ut, eller legge til DNA i kuttsonen, for eksempel bytte ut en sykdomsgivende mutasjon med en frisk DNA-sekvens. Gjengitt fra www.bioteknologiradet.no med tillatelse
En rekke CRISPR-baserte forsøk pågår ved blodsykdommer som hemofili A og B, ved cystisk fibrose, retinitis pigmentosa, muskelsykdommer og kreft, særlig de sjeldne kreftformene som skyldes enkeltgenfeil. Mange degenerative sykdommer i sentralnervesystemet fra barn til sen voksen alder er også gjenstand for slike aktive genterapiforsøk.
Sjeldne sykdommer og behandling
Etter gjeldende definisjon er en sjelden sykdom en tilstand som forekommer med en maksimal kumulativ prevalens på 1 per 2 000 personer. Dette ble i juni 2019 forandret av Helse- og omsorgsdepartementet for å harmonisere med EU-definisjonen fra 1 per 10 000, noe som forandret maksimumstallet i Norge fra vel 500 tilfeller til 2 750 individer.
De fleste av disse sjeldne sykdommene er arvelige. På verdensbasis tilfredsstiller vel 5 000 sykdommer eller tilstander kriteriene, og vel 1000 av disse er registrert i Norge (7). I vårt land rammer mange sykdommer færre enn ti individer for enkeltdiagnoser. Markedet for medikamentutvikling er lite, fordi de fleste av tilstandene er så sjeldne. Den farmasøytiske industrien har derfor hatt en begrenset interesse for feltet, fordi fortjenestemulighetene er potensielt større på andre felter, slik som ved de store folkesykdommene.
Men generell medikamenttørke har sammen med fremskritt i grunnforskningen gitt økt interesse for sjeldenfeltet. Store farmasøytiske konserner kjøper opp mindre bioteknologiske selskaper og utvikler i raskt tempo legemidler som i økende takt bringes på markedet til en meget høy pris. Dette skaper vansker med hensyn til evnen og mulighetene for å kunne gi best tilgjengelige behandling til alle pasienter med sjelden sykdom.
Medikamentene vil ha høye utviklingskostnader. Standardiserte prosedyrer med hensyn til vektorteknologi og klippe-lime-teknikker vil forhåpentligvis bringe kostnadene ned. Markedsføringskrav er krevende, fordi langtids sikkerhetskrav er vanskelig å oppfylle.
Hvilke sykdommer er nå tilgjengelige for genterapi?
I 2020 er syv preparater tilgjengelige, ifølge European Medicines Agency og Food and Drug Administration (1): to preparater mot spinal muskeldystrofi (Spinraza og Zolgensma), ett preparat mot Lebers hereditære optikusnevropati, ett mot betatalassemi og ett mot sigdcelleanemi, samt to preparater mot B-celleleukemi og mantelcellelymfomer, såkalte CAR-T-preparater (chimeric antigen receptor – T-celler), som er en kompleks form for immunterapi.
I det følgende vil jeg belyse noen muligheter og problemer knyttet til genterapi ved å gi en nærmere omtale av spinal muskelatrofi og de to preparatene mot denne tilstanden. Av disse er Spinraza tilgjengelig og i bruk i Norge, mens Zolgensma er til vurdering i Nye metoder per januar 2021(www.nyemetoder.no). De to CAR-T-preparatene er begrenset tilgjengelige for deltagere i forsøk.
Spinal muskelatrofi
Spinal muskelatrofi er recessivt nedarvet. Begge foreldre er bærere, men friske, og tilstanden skyldes en mutasjon i genet på kromosom 5 som gir mangel på survival motor neuron-proteinet (SMN). Alvorlighetsgraden av sykdommen varierer fra symptomer intrauterint eller ved fødselen, i tidlig spedbarnsalder, til senere i barndommen etter at barnet har lært å gå.
Sykdomsdebuten er avhengig av hvor mange kopier pasienten har av SMN2-genet som produserer et lignende genprodukt som ikke er fullgodt, men som gir et protein med nedsatt effektivitet. Ved to og tre kopier kommer symptomene tidlig, ved fire kopier noe senere og sykdomsprogredieringen er da også langsommere. Spinraza øker produksjonen av SMN-proteinet, slik at de motoriske forhornscellene overlever lenger. Som ved poliomyelitt, er imidlertid de døde cellene umulig å vekke til live igjen, så tapt funksjon er stort sett tapt.
Spinraza gis ved injeksjon i spinalkanalen og har relativt kortvarig virkning. Det må settes fire ganger i løpet av første måneder etter start og deretter tre ganger i året. Zolgensma har fordelen av å skulle administreres som en engangsinjeksjon intravenøst. Dette skal deretter ikke være nødvendig å gjenta. Preparatet benytter et adenoassosiert virus som vektor. Denne behandlingen gir åpenbart mindre langtidskostnader til sykehusprosedyrer og oppfølging. Begge preparatene tåles angivelig bra, riktignok med en forbigående økning av leverenzymer ved Zolgensma.
Spinraza er et preparat som baserer seg på oligonukleotidprinsippet. Det lages et såkalt antisense-RNA-molekyl med omkring 20 baser rettet mot feilaktig spleising av et pre-RNA. Zolgensma baserer seg på at hele SMN1-genet settes inn med en virusvektor. Resultatene viser så langt at tidlig behandling gir vesentlig bedre funksjon for begge preparater, men foreløpig er det ikke publisert direkte sammenlignbare resultater som gir grunnlag for å si at det ene preparatet er bedre enn det andre (8).
Den fremforhandlete prisen helseforetakene betaler for Spinraza er hemmelig, men ligger åpenbart langt over fastsatt øvre grense for hva et kvalitetsmessig vunnet leveår vanligvis er verdt i helseøkonomiske regnestykker. For sjeldne sykdommer er det grovt anslått til omkring 900 000 NOK i prioritetsmeldingen (9). Før rabatt koster det første året nær 7 millioner per pasient, deretter 3 millioner årlig. Engangsprisen for Zolgensma er 2,1 millioner amerikanske dollar, som innebærer at preparatet bare unntaksvis er finansiert i offentlige helsesystemer, noen steder i Tyskland og Østerrike, per november 2020. Over 700 pasienter er hittil behandlet med preparatet og det finnes langtidsresultater for de første på over seks år der effekten synes å vedvare, ifølge opplysninger fra produsenten Novartis (6.1.2021).
Huntingtons sykdom som eksempel på hvilke problemer som ligger foran oss
En annen tilstand som belyser muligheter og utfordringer ved genterapi, er Huntingtons sykdom, både med henblikk på ressurser, personalinnsats og kostnader. Jeg har fulgt utviklingen av, eller rettere mangelen på, kausal terapi for denne sykdommen gjennom et langt legeliv.
Sykdommen gir ufrivillige bevegelser, personlighetsforandringer og demens. Den er dominant arvelig og starter oftest i 30-50-årsalderen. De fleste har reprodusert seg når sykdommen debuterer. Det tar lang tid før døden inntreffer, oftest 15-20 år etter symptomdebut, med store belastninger på pasienter, pårørende og helsevesenet som følge.
Det er til enhver tid vel 300 pasienter med Huntingtons sykdom i Norge (10). Siden genfeilen ble lokalisert til kromosom 4 i 1983 og direkte testbar i blod i 1993, har det vært mulig å gjennomføre presymptomatiske tester som viser hvem som vil utvikle sykdom i disse familiene. Imidlertid er det relativt få, om lag 15–20 % av risikopersonene, som har valgt å teste seg. Ut fra forekomsten regner vi at omtrent 600–700 personer er arvebærere. Det pågår tre fase 3-forsøk med antisense-terapi over flere år, alle på flere enn 100 pasienter, behandlet med aktivt preparat hos pasienter i tidlig fase. Det største omfatter nær 800 pasienter som behandles med antisenseoligonukleotider i gjentatte intraspinale injeksjoner i samme prinsipp som Spinraza. Det er et tilsvarende antall i placebogruppene (dobbeltblindforsøk). Studiene er teknisk krevende og kostbare å gjennomføre.
Her hindrer man produksjonen av det proteinet som presumptivt gir patologiske avleiringer i cellekjernen, og som fører til celledød. Fordi sykdommen utvikler seg så langsomt, må man behandle et stort antall pasienter lenge før man kan uttale seg om kliniske effekter. Etter modellforsøk på mus og mennesker er det åpenbart at proteinproduksjonen hemmes og at det neppe er mye bivirkninger på kort sikt.
Dersom preparatet virker i dobbeltblindforsøkene og oppnår markedsføringstillatelse fra legemiddelmyndighetene, gjenstår to betydelige problemer som krever store ressurser personellmessig og finansielt. I tillegg til pasientene med erkjent sykdom er det om lag 700 genbærere, som senere vil utvikle sykdommen. Dersom man skal gjennomføre presymptomatisk testing hos omtrent det dobbelte av antall syke pasienter, er dette en omstendelig prosedyre, fordi de testede trenger tid til å motta og bearbeide prøvesvaret, enten det er godt eller dårlig. Dersom man så skal behandle dem som senere vil utvikle sykdommen, vil dette kreve mye ressurser til medikamenter og injeksjonsprosedyrer. Kostnadsmessig må man anta at dette blir over grensen for hva helsevesenet hittil er rigget til å bære. Siden det åpenbart er viktig å behandle før cellesvinnet er kommet i gang, må behandlingen starte før pasienten får nok symptomer til at man diagnostiserer sykdommen klinisk. Det er vanskelig å si noe om når sykdommen bryter ut, eller rettere lar seg diagnostisere, fordi det er et langt stadium med diffuse symptomer og funn på forhånd. Ved avansert nevropsykologisk og motorisk testing kan man spore sykdomsstart så lenge som 15 år før diagnosen kan stilles.
Bivirkninger og risiko
De første forsøkene med genterapi fra 1990 og utover viste at teknikken var umoden, virusdosen ble for stor og i alle fall to pasienter døde av dette. I mange tilfeller har virkningen vært der, men for kortvarig og i atter andre tilfeller utviklet pasienter som nevnt kreft. Nå synes dette i alle fall foreløpig å være under kontroll. Virkningen av virusvektoren på organismen er imidlertid ikke endelig avklart. Trenger pasientene etter oppstart flere injeksjoner, kan antistoffdannelse bli et problem.
Store gener lar seg foreløpig ikke sette inn i virus. Det gjør at sykdommer som hemofili A, Duchennes muskeldystrofi og nevrofibromatose må angripes med andre metoder som f.eks. CRISPR-teknologi.
Man skiller mellom to typer genterapi: behandling rettet mot somatiske celler og behandling som også kan være rettet mot kjønnsceller, se www.bioteknologiradet.no. At genterapi kan medføre forandringer på kjønnscellene som gjør at genforandringene kan gå i arv, kan ikke utelukkes. Men så lenge det dreier seg om genforandringer for alvorlig sykdom, er dette neppe et så alvorlig onde at det skal avholde en fra behandling. På den annen side kunne dette tenkes å være gunstig f.eks. ved Huntingtons sykdom; da vil neste generasjon også slippe sykdommen.
Avslutning
Noen fantaserer om å bruke genterapi til å forbedre normale egenskaper som idrettsprestasjoner, intelligens, forhindre aldring osv. Den kinesiske forskeren som satte inn et HLA-resistens-gen mot hiv, er imidlertid unisont fordømt og sitter i fengsel for dette. Derimot synes risikoen for toksiske langtidsvirkninger å være uavklart. Observasjonstiden er for kort til at man kan være sikker. Man har mindre enn ti års observasjonstid i de fleste tilfeller, fra de første frivillige ble behandlet og til nå. Ved de sykdommene som jeg har omtalt ovenfor, ja, ved langt de fleste av de genetiske sykdommene som inngår i definisjonen, og særlig de sjeldneste av disse, finnes det ingen effektiv behandling rettet mot grunnårsaken. Det etiske dilemmaet vil være at pasientene ikke har andre alternativer.
Kostnadene vil på sikt sikkert bli det største problemet. Lederen av Bioteknologirådet, Ole Frithjof Norheim, har reist spørsmålet om våre prioriteringskriterier er tilstrekkelige og hvordan nye betalingsordninger kan implementeres i helsevesenet (11). Han peker også på risikoen for et todelt helsevesen dersom vi ikke klarer å finne plass til denne typen behandling i det regulære helsevesenet.
Litteratur
High KA, Roncarolo MG. Gene therapy. N Engl J Med 2019; 381: 455-64. doi: 10.1056/NEJMra1706910.
Genredigering/CRISPR: Teknologien. https://www.bioteknologiradet.no/temaer/genredigering-crispr/genredigeringcrispr-fagressurser-om-teknologien/genredigeringcrispr-teknologien (9.12.2020).
Bratlie S, Kvale H. Nobelprisen til teknologien som endrer alt. Aftenposten 8.10.2020: 38.
Taskén K, Helland Å, Smeland S et al. Nytt initiativ for innføring av persontilpasset kreftbehandling. Aftenposten 3.11.2020: 26-7.
Ribeil JA, Hacein-Bey-Abina S, Payen E et al. Gene therapy in a patient with sickle cell disease. N Engl J Med 2017; 376: 848-55. doi: 10.1056/NEJMoa1609677.
Hirsch T, Rothoeft T, Teig N et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017; 551: 327-32. doi: 10.1038/nature24487.
Opplysninger fra Nasjonal kompetansetjeneste for sjeldne diagnoser (NKSD).
Schorling DC, Pechmann A, Kirschner J. Advances in treatment of spinal muscular atrophy: new phenotypes, new challenges, new implications for care. J Neuromuscul Dis 2020; 7: 1-13. doi:10.3233/JND-190424.
Meld. St. (2015–2016). Verdier i pasientens helsetjeneste – Melding om prioritering. https://www.regjeringen.no/no/dokumenter/meld.-st.-34-20152016/id2502758 (7.1.2021).
Solberg OK, Filkuková P, Frich JC et al. Age at death and causes of death in patients with Huntington disease in Norway in 1986-2015. J Huntingtons Dis 2018; 7: 77-86. doi: 10.3233/JHD-170270.
Norheim OF. Ny gen-terapi utfordrer vår etikk – og vårt prioriteringssystem. Dagens Medisin 3.11.2019. https://www.dagensmedisin.no/artikler/2019/11/03/ny-genterapi-utfordrer-var-etikk--og-vart-prioriteringssystem (7.1.2021).
Arvid Heiberg er spesialist i medisinsk genetikk, overlege og professor emeritus.
Seksjon for klinisk genetikk
Avdeling for medisinsk genetikk
Oslo universitetssykehus
Postboks 4950 Nydalen
0454 Oslo